RNDr. Anna Uhrová Mészárosová, Ph.D.
Vedoucí laboratoře
anna.meszarosova@lfmotol.cuni.cz Genetika hereditárních spastických paraparézNeurogenetická laboratoř Kliniky dětské neurologie 2. LF UK a FNMH (NGL) je vysoce specializované univerzitní a nemocniční laboratorní pracoviště, které spojuje oblast léčebně preventivní péče, výzkumnou i pedagogickou. Laboratoř má více než dvacetiletou tradici a svým zaměřením je unikátní na celorepublikové úrovni. Laboratoř je akreditována dle mezinárodní normy ČSN EN ISO 15189.
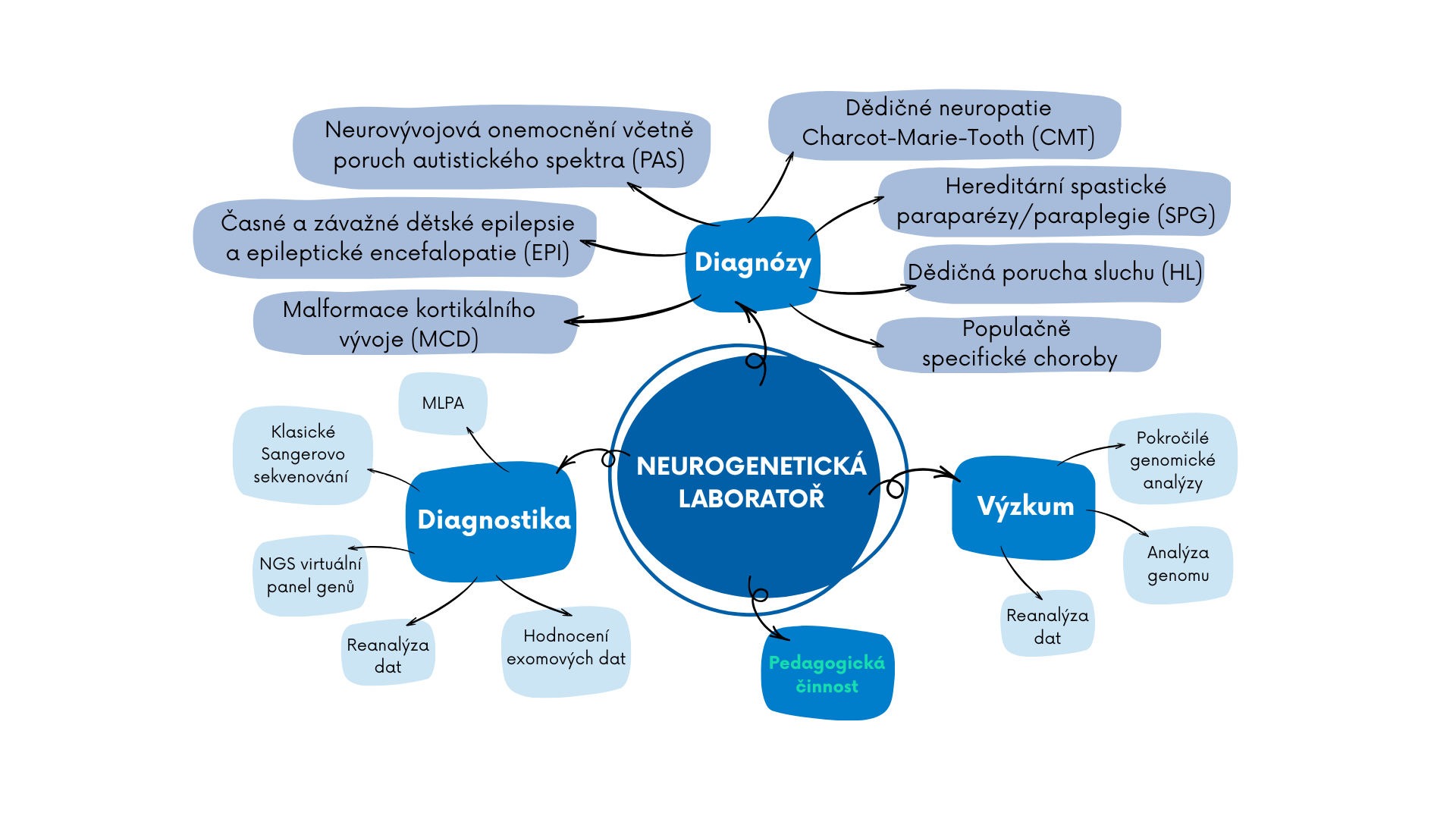
Laboratoř je zaměřena na diagnostiku genetických příčin těchto dědičných neurologických onemocnění: neuropatie Charcot-Marie-Tooth, spastické paraparézy a dalších nervosvalových onemocnění, nesyndromové poruchy sluchu, závažných epilepsií dětského věku, kortikálních malformací a neurovývojových poruch včetně poruch autistického spektra.

Neurogenetická laboratoř
Klinika dětské neurologie 2. LF a FNMH
V Úvalu 84
150 00 Praha 5 – Motol
Telefon: 224 436 788
Umístění: FNMH, část pro dospělé, 3. patro, uzel E

Neurogenetická laboratoř Kliniky dětské neurologie 2. LF UK a FNMH (NGL) je vysoce specializované univerzitní a nemocniční laboratorní pracoviště, které spojuje oblast léčebně preventivní péče, výzkumnou i pedagogickou. Laboratoř má více než dvacetiletou tradici a svým zaměřením je unikátní na celorepublikové úrovni. Zabývá se diagnostikou genetických příčin vzácných dědičných neurologických onemocnění: neuropatie Charcot-Marie-Tooth, spastické paraparézy a dalších nervosvalových onemocnění, nesyndromové poruchy sluchu, závažných epilepsií dětského věku, kortikálních malformací a neurovývojových poruch včetně poruch autistického spektra. Laboratoř je akreditována dle mezinárodní normy ČSN EN ISO 15189.
Laboratoř spolupracuje zejména s klinikami dětské a dospělé neurologie v rámci celé České republiky, poskytuje laboratorní služby pro FNMH a další nemocnice v České republice, v oblasti diagnostiky i výzkumu onemocnění, kterými se zabývá. Dlouhodobě se laboratoř účastní externího hodnocení kvality v rámci mezinárodního systému kontroly kvality The European Molecular Genetics Quality Network (EMQN).
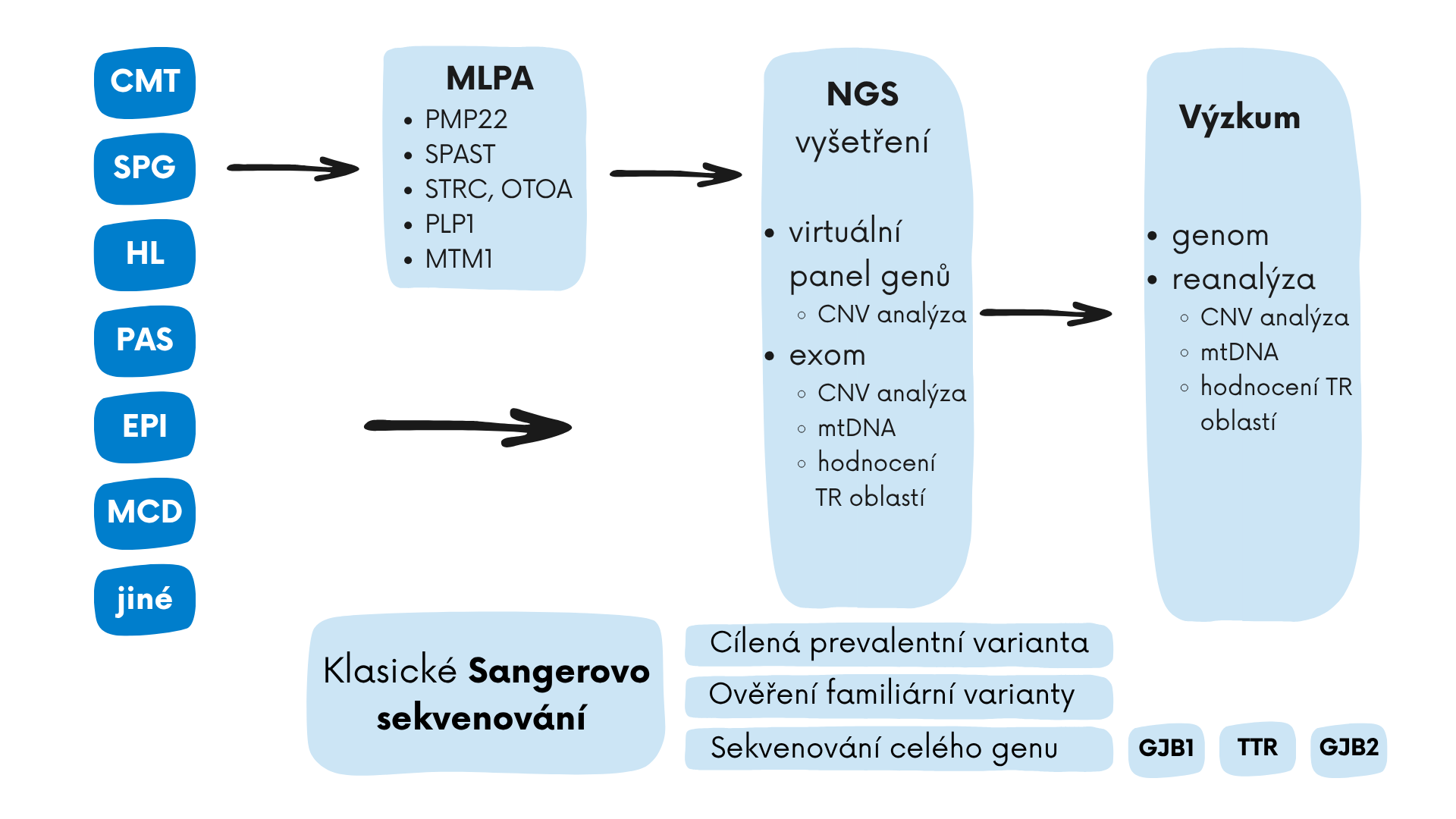
V diagnostice kombinuje laboratoř rutinní i pokročilé genomické metody včetně genomového sekvenování, vyšetření mitochondriálního genomu, analýzu oblastí s expanzemi repetitivních úseků z dat exomového sekvenování. Pacientům a spolupracujícím odborníkům nabízíme kontinuální dlouhodobou diagnostiku s možností rozšíření vyšetření dle individuálních požadavků nebo reanalýzu dat s přihlédnutím k novým poznatkům.
Vědecko-výzkumná činnost představuje dlouhodobě významnou náplň činnosti laboratoře s mnoha výstupy v podobě četných publikací v zahraničních impaktovaných časopisech. Tým laboratoře je úspěšným řešitelem mnoha grantů a členové týmu jsou držitelé řady významných ocenění za publikační a výzkumnou činnost. Dlouhodobě laboratoř spolupracuje s několika významnými zahraničními pracovišti (Rakousko, Německo, USA). Vědecká činnost se úzce prolíná s činností léčebně preventivní a představuje dynamicky se rozvíjející formu propojení výzkumu s praxí. V rámci pedagogické činnosti NGL zajišťuje pregraduální i postgraduální výuku domácích i zahraničních studentů 2. LF UK i dalších fakult.

Vedoucí laboratoře
anna.meszarosova@lfmotol.cuni.cz Genetika hereditárních spastických paraparéz
Zástupce vedoucí
dana.brozkova@lfmotol.cuni.cz Genetika dědičných neuropatií, vrozené nesyndromové hluchoty a vzácných onemocnění romské populace






Pomocí vyšetření DNA lze uvažovanou diagnózu potvrdit, ale v zásadě jí není možné vyloučit. Toto pravidlo platí pro jakékoliv genetické onemocnění. Cesta k nalezení správné diagnózy je mnohdy velmi dlouhá, se souhlasem pacienta proto uchováváme genetický materiál u nás v laboratoři pro další možná vyšetření v budoucnu.
Možnosti vyšetření:
| vyšetření počtu kopií genu metodou MLPA | PMP22 | Charcot-Marie-Tooth choroba, typ l, Tomakulózní neuropatie, HNPP |
| vyšetření NGS panelu genů pro CMT, HMSN, HMN, HSN | CMT panel | Hodnocení virtuálního panelu genů z exomových dat včetně CNV analýzy |
| klasické sekvenování celého kódujícího exonu (úseku) | GJB1 | Charcot-Marie-Tooth choroba, X dominantní |
| klasické sekvenování všech kódujících exonů | TTR | Familiární transtyretinová polyneuropatie |
| klasické sekvenování všech kódujících exonů | PMP22 | Charcot-Marie-Tooth choroba, typ l, Tomakulózní neuropatie, HNPP |
| cílené vyšetření prevalentní patogenní varianty c.442C>T (p.R148*) klasickým sekvenováním | NDRG1 | Charcot-Marie Tooth choroba, typ 4D, CMT4D, HMSN Lom |
| cílené vyšetření prevalentní patogenní intronové varianty c.-249-3818G>C klasickým sekvenováním | HK1 | Charcot-Marie Tooth choroba, typ 4G, CMT4G, HMSN Russe |
Termíny vyhotovení vyšetření:
| delece/duplikace PMP22 genu metodou MLPA | 1-2 měsíce |
| cílené vyšetření klasickým sekvenováním | 1-2 měsíce |
| NGS panelu genů spojených s dědičnými neuropatiemi | 2-4 měsíce |
Dědičné neuropatie CMT neboli také hereditární motorické a senzitivní neuropatie (HMSN) jsou nejčastější dědičná nervosvalová onemocnění, které postihují asi 4 tisíce osob v České republice. CMT se dědí všemi typy mendelovské dědičnosti. Poprvé byla popsána v letech 1856-1868 třemi lékaři (Jean – Marie Charcot, Pierre Marie a Howard Henry Tooth), účinná léčba však dosud není k dispozici.
U pacientů postižených CMT se postupně zhoršuje síla a hybnost dolních a posléze i horních končetin jako následek poškození jejich periferních nervů. CMT není smrtelné onemocnění a ve většině případů nijak nezkracuje očekávanou délku života. Vede však nezřídka k tělesné invalidizaci.
Projevy CMT choroby jsou velice variabilní, a to jak mezi jednotlivými rodinami, tak i v rámci rodiny samotné. CMT choroby jsou způsobovány poruchami velkého počtu genů rozmístěných po celém genomu.
Dědičné neuropatie se klasifikují na základě rychlosti vedení periferním nervem při elektromyografickém vyšetření (EMG) na CMT1 – demyelinizační typ (rychlost vedení nervem medianem pod 38 m/s) a CMT2 – axonální typ (rychlost vedení nad 38 m/s). Proto je pro správnou diagnostiku potřeba elektromyografické (EMG) vyšetření. Další možností je rozdělit dědičné neuropatie na 3 základní skupiny. První a nejčastější – HMSN – dědičné motoricko senzitivní neuropatie, kde je postižen motorický i senzitivní nerv. Druhá skupina dědičných senzitivních neuropatií (HSN), kde jsou výrazněji postiženy senzitivní nervy a třetí skupina dědičných motorických neuropatií (HMN), s větším postižením motorických nervů. Zdaleka nejčastější typ CMT je CMT1A. Je způsoben duplikací 1.4 Mb velké oblasti na chromozomu 17, která obsahuje gen PMP22. Vzácně mohou být příčinou tohoto onemocnění i bodové patogenní varianty v genu PMP22. CMT1A představuje 60-70 % všech neuropatií. Typický nástup obtíží je v 1. dekádě nebo ve školním věku, kdy pacient pozoruje poruchu chůze – zakopávání, případně deformitu nohou – vysoký nárt. Klinicky mírnějším typem CMT je dědičná neuropatie se sklonem k tlakovým parézám tzv. tomakulózní neuropatie (HNPP). Je způsobena delecí 1.4Mb velké oblasti na chromozomu 17. Klinickým projevem jsou opakující se motorické i senzitivní parézy jednotlivých nervů v různých lokalitách. Druhým nejčastějším typem CMT je CMTX1 způsobená kauzálními variantami v genu GJB1, který leží na pohlavním chromozomu X. V tomto případě jsou tedy muži postiženi dříve a více než ženy.
V laboratoři samostatně vyšetřujeme duplikaci/deleci genu PMP22 (nejčastější příčina CMT neuropatie), a dále klasickým (Sangerovým) sekvenováním samostatně vyšetřujeme geny GJB1, TTR (transtyretinová polyneuropatie), PMP22 a dvě konkrétní populačně specifické prevalentní varianty v genu NDRG1 a HK1 (neuropatie typu HMSN Lom nebo HMSN Russe). Dále nabízí laboratoř vyšetření pomocí cíleného panelu vybraných genů spojovaných s dědičnými neuropatiemi. Vyšetření je provedeno pomocí vyhodnocení virtuálního panelu vybraných genů z dat exomového sekvenování (ES). Součástí hodnocení panelu genů je i analýza CNV (copy number variations) zaměřená na detekci velkých delecí nebo duplikací v genech. Vyšetření je dále možné rozšířit po zaslání žádosti na vyhodnocení dat z celého exomu, součástí je také CNV a TR analýza a analýza mitochondriálního genomu (rozšířené vyšetření je prováděno mimo rozsah akreditace).
Možnosti vyšetření:
| Vyšetření NGS panelu genů | EPI panel | Hodnocení virtuálního panelu genů z exomových dat včetně CNV analýzy |
Termíny vyhotovení vyšetření:
| NGS panelu genů spojených s epileptickými encefalopatiemi | 2-4 měsíce |
Epileptické encefalopatie (EE) neboli též závažné časné dětské epilepsie jsou extrémně heterogenní skupinou onemocnění, s časným nástupem epileptických záchvatů, obvykle farmakorezistentních, provázených vývojovou stagnací či regresem. Objasnění příčin, kterých je velmi mnoho u těchto epilepsií, umožnilo až masivně paralelní sekvenování. Pravděpodobnost zjištění genetické příčiny se liší u různých typů epilepsie. U vzácných, závažných, sporadických epilepsií s nástupem v dětském věku s nelezionálním nálezem na magnetické rezonanci mozku je v současnosti až 50% pravděpodobnost, že se pomocí genetických metod (NGS panelu genů) podaří příčinu onemocnění objasnit. Ve většině případů (až 90 %) se jedná o de novo patogenní varianty s autosomálně dominantní dědičností. U běžných, byť často familiárních epilepsií, je tato pravděpodobnost nesrovnatelně nižší.
Laboratoř nabízí vyšetření genů spojovaných s epilepsiemi a EE. Vyšetření je provedeno pomocí vyhodnocení virtuálního panelu vybraných genů z dat exomového sekvenování (ES). Součástí hodnocení panelu genů je i analýza CNV (copy number variations) zaměřená na detekci velkých delecí nebo duplikací v genech. Vyšetření je dále možné rozšířit po zaslání žádosti na vyhodnocení dat z celého exomu, součástí je také CNV a TR analýza a analýza mitochondriálního genomu (rozšířené vyšetření je prováděno mimo rozsah akreditace.
Možnosti vyšetření:
| vyšetření počtu kopií genu metodou MLPA | STRC, OTOA, CATSPER2 | Porucha sluchu: DFNB16, DFNB22 |
| vyšetření NGS panelu genů | HL panel | Hodnocení virtuálního panelu genů z exomových dat včetně CNV analýzy |
| klasické sekvenování všech exonů | GJB2 | Porucha sluchu DFNB1A |
| cílené ověření variant v genu STRC bez přítomnosti pseudogenu STRCP1 | STRC | Porucha sluchu DFNB16 |
| cílené vyšetření prevalentní patogenní varianty c. 2158-2A>G klasickým sekvenováním | MANBA | Beta-mannosidósa – porucha sluchu a mentální retardace |
| cílené vyšetření prevalentní patogenní varianty c.1331+2T>C klasickým sekvenováním. | MARVELD2 | Porucha sluchu typ DFNB49 |
Termíny vyhotovení vyšetření:
| MLPA vyšetření genu STRC, OTOA, CATSPER2 | 1-2 měsíce |
| cílené vyšetření klasickým sekvenováním | 1-2 měsíce |
| NGS panelu genů spojených s dědičnou poruchou sluchu | 2-4 měsíce |
Porucha sluchu je nečastější smyslovou vadou s prevalencí mezi novorozenci 1:800. Více než 60 % případů má genetickou příčinu. Dědičná porucha sluchu je velice různorodá skupina, kde největší zastoupení mají autosomálně recesivně dědičné poruchy sluchu DFNB (75-80 %), které jsou časné a těžší, následované autosomálně dominantními DFNA (20-25 %), které jsou pozdnější a mírnější a X vázanou DNFX (1-1,5 %) poruchou sluchu. V současnosti je známo mnoho genů, jejichž kauzální varianty způsobují dědičnou poruchu sluchu. Až 40 % pacientů s recesivní poruchou sluchu má bialelické patogenní varianty v genu GJB2, jde o typ DFNB1A. Dalších 5-6 % pacientů je objasněno detekcí bialelických patogenní variant, převážně delecí, v genu STRC, pak jde o typ DFNB16.
Laboratoř nabízí vyšetření genu STRC (součástí vyšetření jsou i geny OTOA a CATSPER2) metodou MLPA, který je druhý nejčastěji postižený gen u pacientů s HL. Provádíme i sekvenování genu GJB2 Sangerovým sekvenováním, včetně vyšetření intronové varianty c.-23+1G>A (dříve IVS1+1) v nekódujícím exonu 1. Dále nabízíme vyšetření panelu genů, který obsahuje geny dosud spojované s autosomálně recesivně, dominantně a X- vázanou dědičnou poruchou sluchu. Vyšetření je provedeno pomocí vyhodnocení virtuálního panelu vybraných genů z dat exomového sekvenování (ES). Součástí hodnocení panelu genů je i analýza CNV (copy number variations) zaměřená na detekci velkých delecí nebo duplikací v genech. Vyšetření je dále možné rozšířit po zaslání žádosti na vyhodnocení dat z celého exomu, součástí je také CNV a TR analýza a analýza mitochondriálního genomu (rozšířené vyšetření je prováděno mimo rozsah akreditace). Laboratoř nabízí také vyšetření prevalentních variant, které se vyskytují v romské populaci, a to v genech MARVELD2 a MANBA.
Vzhledem ke složité detekci patogenních variant v genu STRC nabízíme ověření jejich přítomnosti v genu STRC s vyloučením sekvence pseudogenu STRCP1. Z NGS vyšetření nelze jednoznačně určit, zda jsou detekované varianty v genu, či pseudogenu, je to dáno velkou homologií obou oblastí. Proto detekované varianty z NGS vždy ověřujeme Sangerovým sekvenováním z long range PCR, kdy amplifikujeme selektivně pouze sekvenci genu. Jen tak lze s jistotou potvrdit, že prokázaná varianta je přítomna v genu a vysvětluje poruchu sluchu.
Literatura
Markova SP, Brozkova DS, Lassuthova P, Meszarosova A, Krutova M, Neupauerova J, et al. STRC Gene Mutations, Mainly Large Deletions, are a Very Important Cause of Early-Onset Hereditary Hearing Loss in the Czech Population. Genet Test Mol Biomarkers. 2018;22(2):127-34.
Možnosti vyšetření:
| vyšetření počtu kopií genu metodou MLPA | SPAST | Spastická paraplegie 4, autozomálně dominantní způsobená velkými delecemi v genu SPAST |
| vyšetření NGS panelu genů | HSP (SPG) panel | Hodnocení virtuálního panelu genů z exomových dat včetně CNV analýzy |
Termíny vyhotovení vyšetření:
| MLPA vyšetření genu SPAST | 1-2 měsíce |
| NGS panelu genů spojených s hereditárními spastickými paraparézami | 2-4 měsíce |
HSP (SPG) jsou heterogenní skupinou dědičných onemocnění centrálního motoneuronu, klinicky se projevují oboustrannou progredující spasticitou a slabostí dolních končetin. Obtíže postupně progredují a mohou vést až k ztrátě schopnosti samostatné chůze. Symptomy se mohou začít projevovat v kterémkoli věku. Existuje klasifikace na skupinu nekomplikovaných HSP a skupinu tzv. komplikovaných HSP, kdy se krom popsaných příznaků u pacientů vyskytují další klinické znaky, např. postižení horních končetin, ataxie, atrofie částí mozku, atrofie optického nervu, kognitivní deficit, mentální retardace, epilepsie a další. Příčinou nemoci mohou být patogenní varianty v mnoha genech se všemi typy mendelovské dědičnosti. Spojitost s HSP byla dosud popsána u více než 200 genů, u 40-60 % pacientů je příčinou onemocnění patogenní varianta v genu SPAST s autosomálně dominantní dědičností způsobujících onemocnění typ SPG4. Laboratoř nabízí vyšetření genů dosud popsaných s fenotypem HSP (na základě databáze HGMD Profess. a našich dosavadních výsledků). Vyšetření je provedeno pomocí vyhodnocení virtuálního panelu vybraných genů z dat exomového sekvenování (ES). Součástí hodnocení panelu genů je i analýza CNV (copy number variations) zaměřená na detekci velkých delecí nebo duplikací v genech. Vyšetření je dále možné rozšířit po zaslání žádosti na vyhodnocení dat z celého exomu, součástí je také CNV a TR analýza a analýza mitochondriálního genomu (rozšířené vyšetření je prováděno mimo rozsah akreditace).
Dále laboratoř nabízí vyšetření velkých delecí v genu SPAST (součástí kitu pro MLPA je i gen ATL1) metodou MLPA. Velké delece v genu SPAST jsou také popsány jako příčina HSP, nejčastějšího typu SPG4, nicméně jsou spíše jen minoritní příčinou. Toto vyšetření je přínosné v rodinách, kde je zřejmá autozomálně dominantní dědičnost a vyšetření panelu genů je negativní.
Možnosti vyšetření:
| vyšetření NGS panelu genů | PAS panel | Hodnocení virtuálního panelu genů z exomových dat včetně CNV analýzy |
Termíny vyhotovení vyšetření:
| NGS panelu genů spojených s neurovývojovým onemocněním | 2-4 měsíce |
Neurovývojové poruchy jsou poruchy chování a kognitivních funkcí, které vznikají během vývojového období a které zahrnují významné obtíže při osvojování a provádění specifických intelektuálních, motorických, jazykových nebo sociálních funkcí. Předpokládaná etiologie neurovývojových poruch je komplexní a v mnoha jednotlivých případech není známa. Poruchy autistického spektra (PAS) představují skupinu neurovývojových onemocnění, které jsou charakterizovány narušením sociálních interakcí, komunikace a stereotypními, opakujícími se vzorci chování, zájmů a aktivit. Současná prevalence poruch autistického spektra se odhaduje až na 2,5 %. Příčiny vzniku PAS jsou komplexní se zastoupením jak genetických faktorů, tak faktorů vnějšího prostředí, ale mohou být také součástí řady chorob se známou genetickou etiologií. Geneticky jsou velmi heterogenní a mohou být způsobeny jak zděděnými, tak nově vzniklými variantami. V posledním desetiletí byly identifikovány stovky genů, které k onemocnění přispívají. Tyto geny však tvoří pouze 10-20 % případů PAS a pacienti s podobnými patogenními variantami mohou být diagnostikováni na velmi rozdílných úrovních spektra.
Laboratoř nabízí vyšetření skupiny genů spojovaných s neurovývojovými poruchami včetně poruch autistického spektra a poruch vývoje intelektu. Vyšetření je provedeno pomocí vyhodnocení virtuálního panelu vybraných genů z dat exomového sekvenování (ES). Součástí hodnocení panelu genů je i analýza CNV (copy number variations) zaměřená na detekci velkých delecí nebo duplikací v genech. Vyšetření je dále možné rozšířit po zaslání žádosti na vyhodnocení dat z celého exomu, součástí je také CNV a TR analýza a analýza mitochondriálního genomu (rozšířené vyšetření je prováděno mimo rozsah akreditace).
Možnosti vyšetření:
| vyšetření NGS panelu genů | MCD panel | Hodnocení virtuálního panelu genů z exomových dat včetně CNV analýzy |
Termíny vyhotovení vyšetření:
| NGS panelu genů spojených s malformacemi kortikálního vývoje | 2-4 měsíce |
Malformace kortikálního vývoje (malformations of cortical development – MCD) představují širokou skupinu vrozených vad mozku, zejména mozkové kůry, ale i podkorových struktur a mozečku, které se mohou klinicky projevit již od fetálního období s maximem výskytu příznaků v kojeneckém věku. Pacienti s MCD přichází k prvnímu vyšetření pro epileptické záchvaty, opoždění psychomotorického vývoje, poruchy svalového tonu, poruchy krmení, atypický obvod hlavy a další. Zlatým standardem diagnostiky pacientů s MCD je, kromě anamnézy a fyzikálního vyšetření, zejména kvalitní zobrazení pomocí magnetické rezonance (MRI) vyhodnocené zkušeným neuroradiologem. Bilaterální MCD vznikají nejčastěji v důsledku patogenních variant v některém z genů pro cytoskelet, které kódují proteiny odpovědné za pohyb jádra a cytoplasmatických výběžků při migraci neuronů, proteiny fungující jako molekulární motory pro axonální transport a další. Fokální MCD vznikají na podkladě patogenních somatických variant v genech MTOR signální kaskády a jejich regulátorů. Specifickou skupinu představují pacienti s patogenními variantami v genech GATOR1 komplexu (DEPDC5, NPRL2, NPRL3), u nichž se může projevit fokální epilepsie bez nálezu fokální kortikální dysplázie (focal cortical dysplasia – FCD) anebo mohou mít fokální epilepsii a jednoznačnou FCD na MRI – u těchto pacientů se epilepsie často vyskytuje v rodinách. Genetické vyšetření má taktéž velký význam u pacientů s fokální farmakorezistentní epilepsií, kteří přichází k vyšetření v epileptochirurgickém programu. Pomáhá nám odlišit vybrané pacienty s geneticky podmíněnou fokální epilepsií, u nichž resekční epileptochirurgie není indikována, od pacientů s MRI-negativní FCD, kteří naopak z chirurgické léčby epilepsie mohou profitovat.
Laboratoř nabízí vyšetření genů spojovaných s malformacemi kortikálního vývoje. Vyšetření je provedeno pomocí vyhodnocení virtuálního panelu vybraných genů z dat exomového sekvenování (ES). Součástí hodnocení panelu genů je i analýza CNV (copy number variations) zaměřená na detekci velkých delecí nebo duplikací v genech. Vyšetření je dále možné rozšířit po zaslání žádosti na vyhodnocení dat z celého exomu, součástí je také CNV a TR analýza a analýza mitochondriálního genomu (rozšířené vyšetření je prováděno mimo rozsah akreditace.
Možnosti vyšetření:
| cílené vyšetření patogenní varianty c.657del5 a patogenní varianty c.643C>T (p.R215W) klasickým sekvenováním | NBN | Syndrom Nijmegen breakage, NBS |
Termíny vyhotovení vyšetření:
| cílené vyšetření variant klasickým sekvenováním | 1-2 měsíce |
NBS (OMIM 251260), známý také jako syndrom Seemanové II, je vzácný autosomálně recesivně dědičný syndrom chromozomální instability, častěji se vyskytující ve slovanské populaci.
Klinické projevy zahrnují závažnou kongenitální mikrocefalii, prenatální a postnatální růstovou retardaci, humorální a celulární imunodeficienci, opakované respirační infekty se sklonem k bronchiektáziím a vývoj většinou lymforetikulární malignity v dětství.
Charakteristická facies s prominencí středních obličejových partií a nosu, ustupujícím nízkým čelem, retromikrognatií a velkými ušními boltci se vyvíjí po 3. roce věku.
Laboratorní nálezy nízkých imunoglobulinů, chromozomálních zlomů s typickými translokacemi chromozomů 7/14, buněčné i chromozomální hyperradiosenzitivity a radiorezistence syntézy DNA podporují diagnózu NBS. Gen, jehož porucha vede k NBS byl objeven v roce 1998, leží na osmém chromozomu v oblasti 8q21 a byl nazván NBS1 a nověji přejmenován na NBN. Přímá detekce nejčastější patogenní varianty v NBN genu (slovanská c.657del5 delece 5 nukleotidů v pozici 657) umožňuje exaktní diagnózu postnatální i prenatální. Gen kóduje protein nibrin, jehož funkcí je oprava chromozomálních zlomů po ionizačním záření.
U homozygotů se chybějící nibrin projeví poruchou stability DNA (DNA repair disorder) vedoucí k malignímu procesu. Hyperradiosenzitivita homozygotů má klinické důsledky pro vznik a léčbu malignit a je důvodem k časné diagnostice a zavedení účinné prevence ochranou před ionizací.
Frekvence přenašečů varianty c.657del5, predominantně se vyskytující ve slovanské populaci, byla nalezena mezi 1:90 v Polsku – Novy Sad po 1:314 v Krakově. Mezi českými novorozenci, narozenými před 20 lety, byla zjištěna frekvence heterozygotů 1:128-154. Testování varianty se provádí u pacientů s mikrocefalií, v rodinách se zvýšeným výskytem malignit, i u např. pracovníků na rizikových pracovištích (RTG). Včasná diagnóza je zásadní pro odpovídající preventivní péči a terapii. U pacientů s NBS je třeba se důsledně chránit před ionizujícím zářením (RTG zářením a dalšími mutageny). V případě rozvoje malignity, což je u pacientů s NBS bohužel velice pravděpodobné, je třeba léčit sníženou dávkou chemoterapie a vyvarovat se použití radiomimetických cytostatik.
Literatura
Seeman P, Gebertova K, Paderova K, Sperling K, Seemanova E. Nijmegen breakage syndrome in 13% of age-matched Czech children with primary microcephaly. Pediatr Neurol. 2004;30(3):195-200.
Možnosti vyšetření:
| vyšetření počtu kopií genu metodou MLPA | PLP1 | Pelizaeus Merzbacherova choroba, PMD |
| klasické sekvenování všech kódujících exonů | PLP2 | Pelizaeus Merzbacherova choroba, PMD |
Termíny vyhotovení vyšetření:
| vyšetření genu metodou MLPA | 1-2 měsíce |
| cílené vyšetření variant klasickým sekvenováním | 1-2 měsíce |
PMD je X vázané onemocnění centrálního nervového systému projevující se poruchou myelinizace CNS (OMIM 312080). Způsobují jej varianty v PLP1 genu ležícím na dlouhém raménku X chromozomu v oblasti Xq22.2.
PLP1 gen může být postižen několika typy mutací. Nejčastější mutací je duplikace genu i jeho přilehlé oblasti a vzácně i triplikace či vyšší počet kopií PLP1 genu. Vzácnější příčinou jsou bodové kauzální varianty PLP1 genu a delece celého genu, která byla popsána zatím pouze velmi sporadicky. Projevy nemoci jsou nystagmus od narození (přítomný u všech pacientů), hypotonie, opoždění psychomotorického vývoje, spastická kvadruparéza, a později u sedících ataxie, tremor a difuzní leukoencefalopatie na MRI snímcích. Rozlišuje se několik forem nemoci, podle závažnosti projevů. Od nejlehčích přes klasickou formu nemoci až po velmi těžkou konnatální. Pacienti postižení lehkou formou nemoci nemívají zkrácenou délku života, pacienti s klasickou formou nemoci se dožívají někdy až šesté dekády věku a pacienti postiženi nejtěžší formou nemoci umírají v první dekádě života. Těžká forma nemoci (konnatální) je typicky způsobena missense variantami a dalšími bodovými variantami v silně konzervovaných oblastech PLP1 genu. Méně nebezpečná forma nemoci (lehčí spastická paraparéza) je způsobena variantami, které neumožní tvorbu PLP proteinu – tzv. null syndrom. Nejčastější patogenní varianta (duplikace PLP1 genu) způsobuje klasickou formu nemoci.
PMD se dědí X recesivním způsobem, kdy postiženi jsou převážně chlapci, ženy jsou ve většině případů nemanifestní přenašečky. U lehčí formy PMD – tzv. “PLP1null syndromu“ je mírnější postižení u chlapců a je přítomna i periferní neuropatie, která normálně není součástí PMD. V rodinách lehčeji postižených pacientů s tzv. PLP1null syndromem se někdy vyskytují ženy u kterých se v dospělosti manifestují projevy spastické paraparézy typ 2 (SPG2) na rozdíl od rodin s těžkým průběhem nemoci u chlapců na podkladu „těžké patogenní varianty“, kde může být u dívek v útlém věku postižení charakteru PMD, které se postupně upravuje.
V laboratoři testujeme jak nejčastější příčinu nemoci, tak bodové varianty PLP1 genu. DNA vyšetření má sloužit k potvrzení klinické diagnosy. Vyloučení PMD pomocí DNA vyšetření v zásadě možné není. K testování mutací přistupujeme až po studiu klinických a genealogických dat, která jsou pro nás tudíž stěžejní při rozhodování. U rodiny pacienta se známou patogenní variantou je možné v případě zájmu provést prenatální vyšetření. Také je možné v rodinách pacienta se známou patogenní variantou testovat ženy v riziku na přítomnost duplikace.
Literatura
Seeman P, Kršek P, Náměstková K, Malíková M, Belšan T, Prošková M. Pelizaeus Merzbacherova choroba (PMD) – detekce nejčastější mutace proteolipid proteinu genu u českých pacientů a rodin s klasickou formou PMD. Čes a Slov Neurol Neurochir. 2003;66/99(2):95-104.
Taube JR, Sperle K, Banser L, Seeman P, Cavan BC, Garbern JY, et al. PMD patient mutations reveal a long-distance intronic interaction that regulates PLP1/DM20 alternative splicing. Hum Mol Genet. 2014;23(20):5464-78.
Možnosti vyšetření:
| vyšetření počtu kopií genu metodou MLPA | MTM1, MTMR1 | X – vázaná myotubulární myopatie |
Termíny vyhotovení vyšetření:
| vyšetření genu metodou MLPA | 1-2 měsíce |
| vyšetření klasickým sekvenováním | 1-2 měsíce |
X – vázaná myotubulární myopatie je závažné onemocnění, které je způsobeno patogenními variantami v genu MTM1 (též. MTMR1). Onemocnění je zapříčiněno mutacemi v genu pro myotubularin 1 (MTM1). Ve většině případů (více než 80 %) se onemocnění prezentuje v novorozeneckém období velmi závažnou hypotonií, hypo – až areflexií a různými stupni respirační insuficience. Mezi hlavní klinické znaky patří novorozenecká hypotonie, novorozenecké respirační selhávání, svalová slabost, snížený objem svalů, porodní délka a obvod hlavy nad 90.percentilem, dlouhé prsty na rukou i nohou, kryptorchismus. Většina dětí umírá v perinatálním období, i když byli popsáni i pacienti přežívající do období dětství. Mentální vývoj je u přežívajících pacientů normální. Vzhledem k X-vázané dědičnosti jsou postižení jedinci mužského pohlaví. Ženy jsou heterozygotní přenašečky a klinicky jsou asymptomatické nebo jen mírně manifestující. Patogenní varianty včetně delecí v genu MTM1 vedou ke ztrátě funkce proteinu, což způsobuje poruchu svalového vyzrávání a svalová vlákna zůstanou ve svém vývoji zastavena ve fetálním stadiu.
Literatura
Laššuthová P, Sebroň V, Zámečník J, Haberlová J, Baxová A, Seeman P: X-vázaná myotubulární myopatie u dvou bratrů v důsledku nové mutace v MTM1 genu – kazuistiky. Cesk Slov Neurol N 2013; 76/109(2): 241-245.
Možnosti vyšetření:
| cílené vyšetření prevalentní patogenní varianty c.442C>T (p.R148*) klasickým sekvenováním | NDRG1 | Charcot-Marie Tooth choroba, typ 4D, CMT4D, HMSN Lom |
| cílené vyšetření prevalentní patogenní intronové varianty c.-249-3818G>C klasickým sekvenováním | HK1 | Charcot-Marie Tooth choroba, typ 4G, CMT4G, HMSN Russe |
| cílené vyšetření prevalentní patogenní varianty c. 2158-2A>G klasickým sekvenováním | MANBA | Beta-mannosidósa – porucha sluchu a mentální retardace |
| klasické sekvenování všech kódujících exonů | MANBA | Beta-mannosidósa – porucha sluchu a mentální retardace |
| cílené vyšetření prevalentní patogenní varianty c.1331+2T>C klasickým sekvenováním. | MARVELD2 | Porucha sluchu typ DFNB49 |
| klasické sekvenování všech kódujících exonů | MARVELD2 | Porucha sluchu typ DFNB49 |
| cílené vyšetření prevalentní patogenní intronové varianty c.863+389C>T (IVS6 + 389 C>T) klasickým sekvenováním | CTDP1 | Kongenitální katarakta s faciální dysmorfií a neuropatií, CCFDN |
| cílené vyšetření prevalentní patogenní varianty c.92G>C, p.(Gly31Ala) klasickým sekvenováním | EXOSC3 | Pontocerebelární hypoplazie typ 1, PCH1 |
| klasické sekvenování všech kódujících exonů | EXOSC3 | Pontocerebelární hypoplazie typ 1, PCH1 |
Termíny vyhotovení vyšetření:
| vyšetření konkrétní varianty klasickým sekvenováním | 1-2 měsíce |
Romové jsou geneticky izolovaná, endogamní menšina, která od 10. století migrovala z Indie do Evropy. V Evropě žije 10–12 milionů Romů a menší populace se nacházejí v Severní Americe a Číně. Jejich endogamie a příbuzenské sňatky vedly k vyšší frekvenci vzácných autozomálně recesivních poruch, z nichž některé jsou pro romskou populaci unikátní a v homozygotním stavu jsou příčinou závažných vrozených onemocnění.
Literatura
Shauna Quinn, Nicola Walsh, Ioana Streata, Athina Ververi, Samarth Kulshrestha, Ratna Dua Puri, Anca Lelia Riza, Aoibhinn Walsh, Kathleen Gorman, Ellen Crushell, Andrew Green, Janna Kenny, Sally Ann Lynch. Catalogue of inherited autosomal recessive disorders found amongst the Roma population of Europe. Eur J Med Genet. 2025;02:73.
Možnosti vyšetření:
| cílené vyšetření prevalentní patogenní intronové varianty c.863+389C>T (IVS6 + 389 C>T) klasickým sekvenováním | CTDP1 | Kongenitální katarakta s faciální dysmorfií a neuropatií, CCFDN |
Termíny vyhotovení vyšetření:
| cílené vyšetření variant klasickým sekvenováním | 1-2 měsíce |
V roce 1999 byla popsána nová klinická jednotka u Romů v Bulharsku. Kromě neuropatie se vyznačuje dalšími typickými rysy, jako jsou kongenitální katarakta, faciální dysmorfismus, kognitivní deficit a podobně. Z anglického názvu se pro skupinu odvodil název CCFDN (OMIM 604168).
Klinický obraz CCFDN je charakterizován komplexní vývojovou poruchou postihující primárně nervový systém. Onemocnění se nejdříve a vždy projeví kongenitální kataraktou, mikrokorneou a mikroftalmem. V dalším průběhu je patrné opoždění motorického i mentálního vývoje. Stigmatizace facies je nápadná až během dětství, projevuje se prominující střední partií obličeje, zduřením měkkých tkání rtů a mikrognácií.
Neuropatie je progresivní, distálního typu, převážně motorická a nejdříve postihuje dolní končetiny. Během dětství a adolescence se přidává i postižení horních končetin i páteře (skolióza, většinou výrazná) a nemoc postupně vede až ke skeletálním deformitám.
Postižení CNS se projevuje mírným až středním, neprogresivním kognitivním deficitem. U některých nemocných je lehká chorea, ataxie a tremor horních končetin. Dalším rysem CCFDN je menší vzrůst, hypogonadismus, případně i sekundární amenorea.
Syndrom CCFDN byl dosud pozorován výhradně u romské populace a pouze v důsledku této jediné patogenní varianty. U CCFDN je autozomálně recesivní typ dědičnosti. Metodou genetického homozygotního mapování byla příčina syndromu lokalizována na osmnáctý chromozom do oblasti 18q23. Později byla identifikována i kauzální varianta c.863+389C>T v CTDP1 genu. Leží v intronu za 6. exonem a ovlivňuje správný sestřih mRNA. V romském etniku se předpokládá efekt zakladatele.
Testování varianty se provádí u pacientů s klinickými příznaky CCFDN (vrozená katarakta, opoždění psychomotorického vývoje, romské etnikum).
Literatura
Lassuthova P, Siskova D, Haberlova J, Sakmaryova I, Filous A, Seeman P. Congenital cataract, facial dysmorphism and demyelinating neuropathy (CCFDN) in 10 Czech Gypsy children–frequent and underestimated cause of disability among Czech Gypsies. Orphanet J Rare Dis. 2014;9:46.
Ke genetickému vyšetření přijímáme tyto typy vzorků:
Vzorky jsou přijímány spolu s žádankou a informovaným souhlasem pacienta (viz. Dokumenty ke stažení). Všechny typy přijímaných vzorků je možné uchovávat až do okamžiku doručení při pokojové teplotě (lépe v lednici). Příjem biologického materiálu probíhá v pracovní dny v provozní době laboratoře prostřednictvím běžné pošty nebo osobně pověřeným zdravotnickým pracovníkem. Vzorky je třeba řádně zabezpečit proti mechanickému poškození a kontaminaci. Laboratoř sama nezajišťuje odběr biologického materiálu.
 Provozní doba laboratoře: Po – Pá 8:00 – 15:30
Provozní doba laboratoře: Po – Pá 8:00 – 15:30
Telefon: 22 443 6788
Adresa:
Neurogenetická laboratoř
Klinika dětské neurologie 2. LF UK a FNMH
V Úvalu 84
150 00 Praha 5 – Motol
Umístění: FNMH, část pro dospělé, 3. patro, uzel E